单细胞数据_scRNASeq 分析一条龙(入门)
工具准备
- R语言:Linux下无root权限安装R语言(conda安装和普通安装)
- python:conda安装
- cellranger:后面用到会说
单细胞技术介绍
转自:https://shixiangwang.github.io/posts/2023-07-13-fundamentals-of-scrnaseq-analysis
技术流程
单细胞RNA测序(scRNA-seq)包括一系列技术,以产生许多单个细胞的全基因组表达数据。
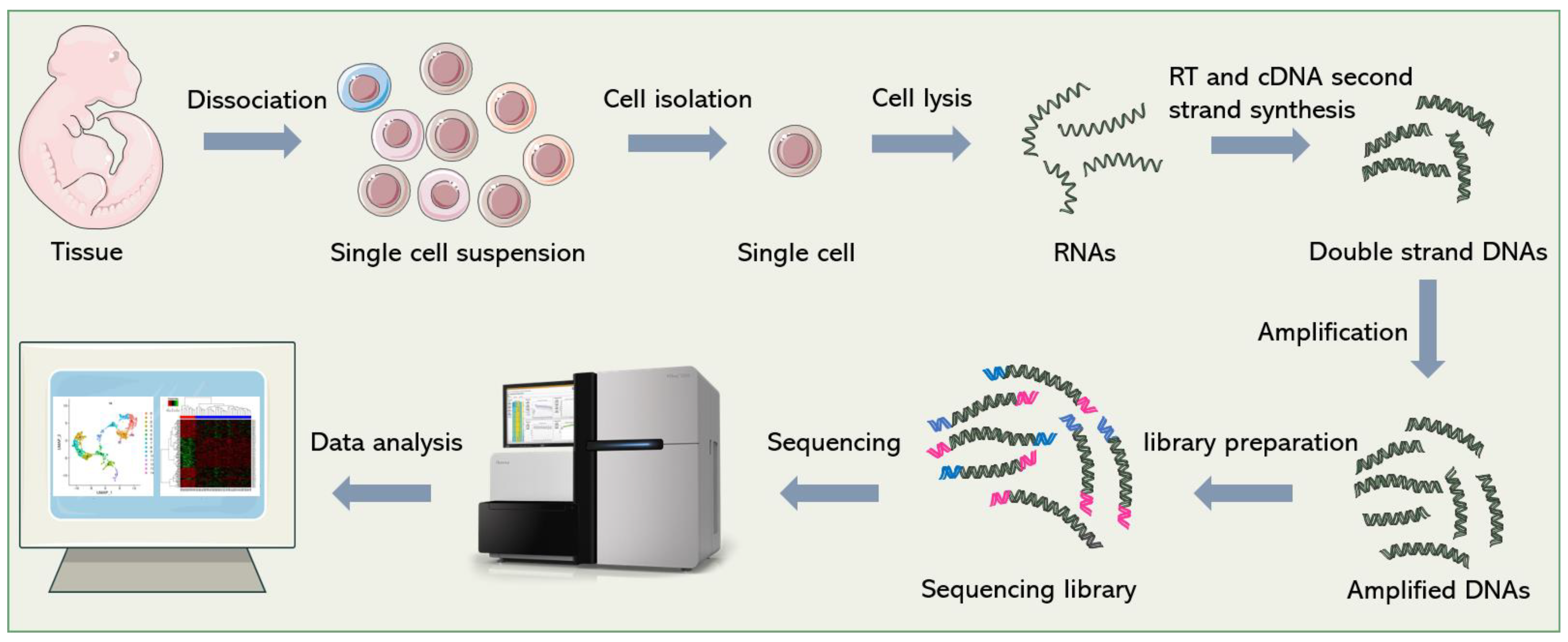
细胞分离
根据技术主要可以分为两类:Droplet-based 和 Non droplet-based。
Droplet-based:
- 组织样品必须解离成悬浮液
- 细胞将被单独封装成一个油包水滴
- 高通量,低成本
- 相关技术:Drop-seq (Macosko et al. 2015), inDrop (Klein et al. 2015), Chromium 10X (Zheng et al. 2017)
Non droplet-based:
- Smart-seq2 (Ramsköld et al. 2012):用微型毛细管移液器手工细胞取样
- CEL-seq (Hashimshony et al. 2012):单个细胞被添加到试管中;第一个介绍了条形码和RNA聚合
- MARS-seq (Jaitin et al. 2014)是第一个使用FACS将单细胞分离到单孔中的方法,优化版MARS-seq2 (Keren-Shaul et al. 2019)的推出成本更低,可重复性更高,井间污染更少
下图显示了一些常见的单细胞分离技术(Hwang, Lee, and Bang 2018)
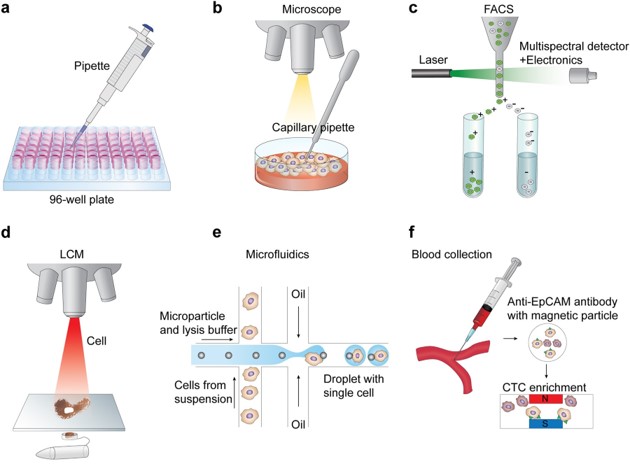
Single Cell Isolation (modified from Hwang, Lee, and Bang 2018)
条形码和唯一分子标识符(UMI)
双端测序输出两个 fastq 文件,分别对应测序的5′和3′方向。使用这种测序技术,配对的第一个read总是与引物的细胞(条形码+UMI)部分一致。

Biased paired-end reads (David Tse et.al)
根据获得的细胞条形码、UMI 和 cDNA 的 reads,我们可以估计转录物的丰度。这允许 mapping 算法区分哪些序列是条形码,哪些是转录序列。因此,在确定细胞条形码和 UMI 条形码序列的长度和位置时,识别用于测序的文库制备化学方法非常重要。
为了获得 UMIs 的计数,我们可以首先通过细胞条形码对 reads 进行分组,然后对齐 cDNA reads 并使用 UMIs 对每个细胞每个基因的独特分子进行计数。
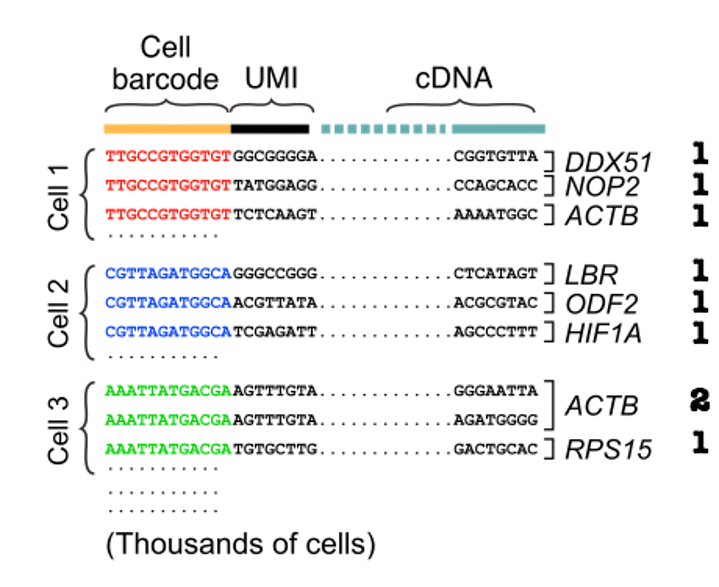
Grouping barcodes to assign reads to cells (modified from David Tse et.al)
对细胞条形码和 UMIs 的分析包括在校准过程中,我们将在下一节中介绍更多内容。
广泛应用的scRNA-seq技术汇总
| Methods | Transcript coverage | UMI possibility | Strand specific | References |
|---|---|---|---|---|
| Tang method | Nearly full-length | No | No | Tang et al. (2009) |
| Quartz-Seq | Full-length | No | No | Sasagawa et al. (2013) |
| SUPeR-seq | Full-length | No | No | X. Fan et al. (2015) |
| Smart-seq | Full-length | No | No | Ramsköld et al. (2012) |
| Smart-seq2 | Full-length | No | No | Picelli et al. (2013) |
| MATQ-seq | Full-length | Yes | Yes | Sheng et al. (2017) |
| STRT-seq STRT/C1 | 5′-only | Yes | Yes | Islam et al. (2011) |
| CEL-seq | 3′-only | Yes | Yes | Hashimshony et al. (2012) |
| CEL-seq2 | 3′-only | Yes | Yes | Hashimshony et al. (2016) |
| MARS-seq | 3′-only | Yes | Yes | Jaitin et al. (2014) |
| CytoSeq | 3′-only | Yes | Yes | H. C. Fan, Fu, and Fodor (2015) |
| Drop-seq | 3′-only | Yes | Yes | Macosko et al. (2015) |
| InDrop | 3′-only | Yes | Yes | Klein et al. (2015) |
| Chromium | 3′-only | Yes | Yes | Zheng et al. (2017) |
| SPLiT-seq | 3′-only | Yes | Yes | Rosenberg et al. (2018) |
| sci-RNA-seq | 3′-only | Yes | Yes | Cao et al. (2017) |
| Seq-Well | 3′-only | Yes | Yes | Gierahn et al. (2017) |
| DroNC-seq | 3′-only | Yes | Yes | Habib et al. (2017) |
| Quartz-Seq2 | 3′-only | Yes | Yes | Sasagawa et al. (2018) |
FASTQ 文件
原始 RNA 测序数据大概率在 FASTQ 文件中。它是一种基于文本的格式,用于存储由单字母代码表示的读序列。 FASTQ 文件中的序列以@符号开头的readID开始,然后是序列数据行,一个简单的加号+分隔符和碱基质量分数。
它以以下格式表示:
@ReadID
READ SEQUENCE
+
SEQUENCING QUALITY SCORES
一般来说,fastq 文件是使用质量控制工具(如 FastQC)进行预处理的。这将输出一系列评估序列 reads 质量的指标。
下载fastq文件
数据库介绍
代码下载fastq数据
Browsing NCBI with rentrez 使用rentrez访问NCBI数据库
安装Toolkits,使用prefetch下载SRA数据库的fastq数据
分析fastq数据(->10x文件 / h5ad文件)
一般不会对fastq文件进行分析,需要先通过软件将fastq文件转化为10x文件/ h5ad文件,一般常用的是Cell ranger,后面会讲解使用教程。其他工具还有:STARsolo,Doublets。
使用Cell ranger分析单细胞数据
Cell Ranger 是一组分析管道,用于处理 Chromium 单细胞数据以对齐 reads,生成特征条形码矩阵,执行聚类和其他二次分析等等。 它帮助我们生成 RNA reads 计数矩阵,我们将在学习中使用。
一些概念:
- GEM 孔(以前称为 GEM 组):来自单个 10x Chromium™ 芯片通道的分隔单元(凝胶颗粒悬浮液)集合。可以从一个 GEM 孔中获得一个或多个测序文库。
- 文库(或测序文库):从单个 GEM 孔中制备的带有 10x 条形码的测序文库。借助特征条形码或 V(D)J 分析,可以从同一个 GEM 孔中创建多个文库。文库类型可能包括基因表达、抗体捕获、CRISPR 引导捕获、TCR 富集等。
- 测序 Run(或 Flowcell): A flowcell containing data from one sequencing instrument run.(这个从英文直译上很难理解,通俗的说就是一次上机测序得到的数据流)
详细使用教程: 使用Cell ranger分析单细胞数据
STARsolo,Doublets简单介绍
转自:https://shixiangwang.github.io/posts/2023-07-13-fundamentals-of-scrnaseq-analysis
STARsolo
STARsolo(Kaminow,Yunusov和Dobin 2021)是一个专为液滴式单细胞RNA测序数据(例如10X Genomics Chromium系统)设计的分析工具,直接内嵌在STAR代码中。STARsolo的输入是一个FASTQ文件,它可以以与Cell Ranger几乎相同的格式输出基因计数,但速度约快10倍。
STARsolo 程序输出大量反映 reads 比对过程细节的文件。在这里,我们只讨论其中一些关键文件。
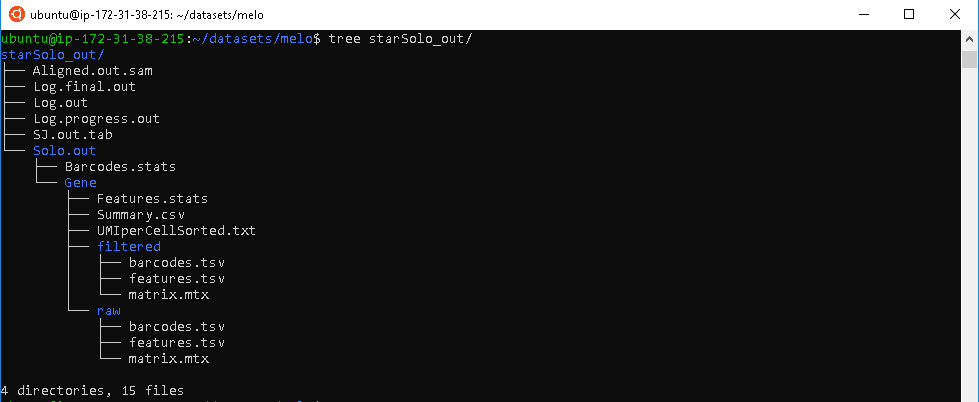
STARsolo Output
- BAM文件包含有关比对 reads 的信息,与通过Cell Ranger生成的BAM文件非常相似。
- 比对摘要文件Features.stats和Summary.csv包含有关基本比对细节的信息。这可以作为对比对过程进行简单初步质量控制检查的便捷工具。
- 特征矩阵文件matrix.mtx包含每个个体细胞中映射的基因计数信息。列名对应于每个个体细胞的条形码,行名对应于所有注释的基因。由于该数据的规模较大,它以稀疏矩阵的形式存储。
- 辅助文件 barcodes.tsv 和 features.tsv 提供了下游分析所需的额外元数据。这些文件连同矩阵文件在功能上扮演着与Cell Ranger输出中的 Matrices 部分相同的角色,并且在金标准的 scRNA-seq 数据分析软件 seurat 中进行分析时是必需的。
Doublets
双细胞是指虽然设计为由一个细胞生成,但却是由两个细胞生成的人工文库。通常这是由于细胞分选或捕获过程中的错误引起的。
可以使用几种实验策略来去除双细胞:
- 基于不同供体个体之间的自然遗传变异(Kang等人,2018)。
- 使用与不同寡核苷酸结合的抗体标记一组细胞(例如,来自一个样本的所有细胞)。在混合后,观察到具有不同寡核苷酸的文库被视为双细胞并移除(Bach等人,2017)。
- 仅基于表达谱的计算方法(如模拟)推断双细胞并进行去除。
质量控制
Python使用Scanpy计算质量控制指标
scanpy计算n_genes_by_counts和total_counts等质量控制指标
其他教程:
-
Python 单细胞分析教程(一):质量控制
https://cloud.tencent.com/developer/article/2320174 -
单细胞测序最好的教程(十):万能的Transformer与细胞注释
https://cloud.tencent.com/developer/article/2326642 -
基于python的单细胞数据预处理-降维可视化
https://blog.csdn.net/qq_40943760/article/details/138717289 -
基于python的单细胞数据预处理-质量控制
https://blog.csdn.net/qq_40943760/article/details/138585941
R语言使用Seurat 进行分析
网上有很多,推荐一个:
使用 Seurat 进行分析:
https://shixiangwang.github.io/posts/2023-07-13-fundamentals-of-scrnaseq-analysis/#使用-seurat-进行分析